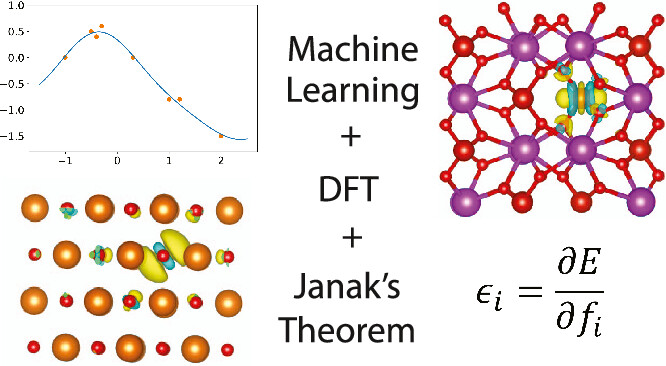
One of the core challenges in semilocal density functional theory (DFT) is the consistent underestimation of band gaps, primarily due to self-interaction and delocalization errors. This issue complicates the prediction of electronic properties and charge transfer mechanisms. Hybrid DFT, incorporating a fraction of exact exchange energy, offers improved band gap predictions but often requires system-specific tuning. Machine learning approaches have emerged to enhance DFT accuracy, particularly for molecular reaction energies and strongly correlated systems. Explicitly fitting energy gaps, as demonstrated by the DM21 functional, could improve DFT predictions by addressing self-interaction errors.
Researchers from Harvard SEAS have developed a machine learning method using Gaussian processes to enhance the precision of density functionals for predicting energy gaps and reaction energies. Their model integrates nonlocal features of the density matrix to forecast molecular energy gaps accurately and estimate polaron formation energies in solids, even though it was trained exclusively on molecular data. This advancement builds upon the CIDER framework, which is known for its efficiency and scalability in handling large systems. Although the model currently targets exchange energy, it holds promise for broader applications, including predicting electronic properties such as band gaps.
The study introduces key concepts and methods for fitting exchange-correlation (XC) functionals in DFT, focusing on band gap prediction and single-particle energies. It covers the application of Gaussian process regression to model XC functionals, incorporating training features that enhance accuracy. The theoretical foundation is based on Janak’s theorem and the concept of derivative discontinuity, which are crucial for predicting properties like ionization potentials, electron affinities, and band gaps within generalized Kohn-Sham DFT. The approach aims to improve functional training by addressing the challenges of orbital occupation and derivative discontinuities.
The CIDER24X exchange energy model was developed using a Gaussian process, enhancing flexibility. Key features were chosen for minimal covariance, adjusted to a specific range, and employed to train a dense neural network approximating the Gaussian process. The training data comprised uniform electron gas exchange energy, molecular energy differences, and energy levels sourced from databases such as W4-11, G21IP, and 3d-SSIP30. Two variants of the CIDER24X model were created: one incorporating energy level data (CIDER24X-e) and another excluding it (CIDER24X-ne) to evaluate the effect of including energy levels in the fitting process.
The study introduces the CIDER24X model with SDMX features, showing enhanced predictive accuracy for molecular energies and HOMO-LUMO gaps compared to previous models and semilocal functionals. CIDER24X-ne, trained without energy levels, aligns closely with PBE0, while CIDER24X-e, which includes energy level data, offers a better balance between energy and band gap predictions. Though trade-offs exist, particularly with eigenvalue training, CIDER24X-e outperforms semilocal functionals and approaches hybrid DFT accuracy, making it a promising alternative that reduces computational costs.
In conclusion, The study presents a framework for fitting density functionals to both total energy and single-particle energy levels using machine learning, leveraging Janak’s theorem. A new set of features, SDMX, is introduced to learn the exchange function without requiring the full exchange operator. The model, CIDER24X-e, maintains accuracy in molecular energies while significantly improving energy gap predictions, matching hybrid DFT results. The framework is extendable to full XC functionals and other machine learning models, offering the potential for efficient and accurate electronic property predictions across diverse systems.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. If you like our work, you will love our newsletter..
Don’t Forget to join our 50k+ ML SubReddit
Here is a highly recommended webinar from our sponsor: ‘Building Performant AI Applications with NVIDIA NIMs and Haystack’
The post Harvard Researchers Introduce a Machine Learning Approach based on Gaussian Processes that Fits Single-Particle Energy Levels appeared first on MarkTechPost.