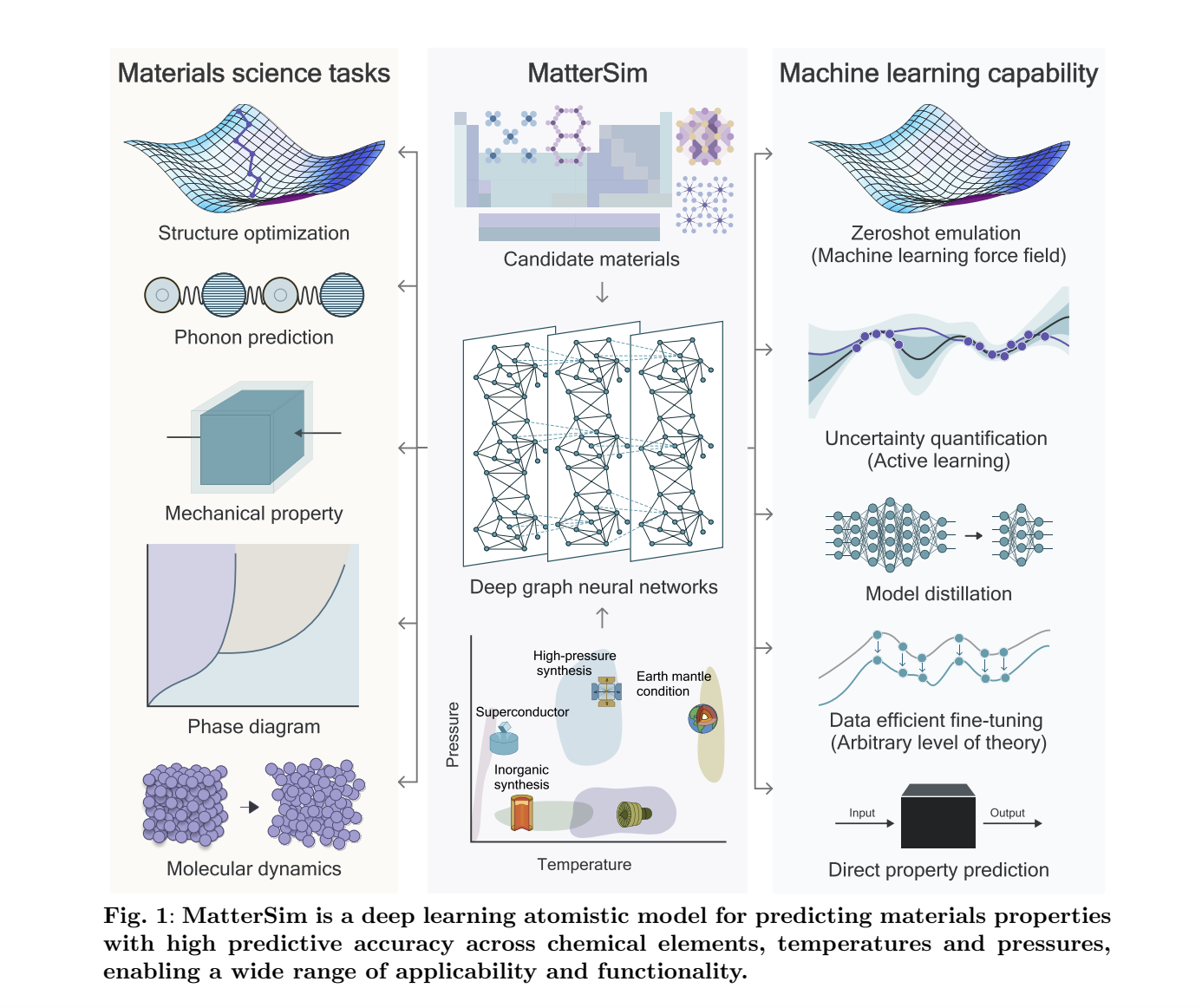
Microsoft has released MatterSimV1-1M and MatterSimV1-5M on GitHub, cutting-edge models in materials science, offering deep-learning atomistic models tailored for precise simulations across diverse elements, temperatures, and pressures. These models, designed for efficient material property prediction and atomistic simulations, promise to transform the field with unprecedented speed and accuracy. MatterSim models operate as a machine learning force field, enabling researchers to simulate and predict the properties of materials under realistic thermodynamic conditions, such as temperatures up to 5000 K and pressures reaching 1000 GPa. Trained on millions of first-principles computations, these models provide insights into various material properties, from lattice dynamics to phase stability.
Material discovery and design were slow, and expensive experimental methods dominated trial-and-error processes. MatterSim models offer an in silico alternative, expediting the prediction and analysis of material properties. Deep learning bridges gaps in traditional ways like Density Functional Theory (DFT), providing faster and comparably accurate results. MatterSim models have been actively developed to simulate materials under diverse conditions. MatterSimV1-1M is trained on one million data points optimized for general-purpose simulations. MatterSimV1-5M, trained on five million data points, provides enhanced accuracy for complex materials and intricate configurations.
MatterSim models accurately predict properties such as Gibbs free energy, mechanical behavior, and phase transitions. Compared to previous best-in-class models, it achieves up to a ten-fold improvement in predictive precision, with a mean absolute error (MAE) as low as 36 meV/atom on datasets covering extensive temperature and pressure ranges. One of the model’s standout features is its capability to predict temperature- and pressure-dependent properties with near-first-principles accuracy. For instance, it accurately forecasts Gibbs free energies across various inorganic solids and computes phase diagrams at minimal computational cost. The model’s architecture integrates advanced deep graph neural networks and uncertainty-aware sampling, ensuring robust generalizability. With active learning, MatterSim models enrich its dataset iteratively, capturing the underrepresented regions of the material design space.
MatterSimV1-1M and MatterSimV1-5M Models excel in several applications:
- Materials Design: It predicts ground-state material structures and energetics, helping researchers discover and refine materials with specific properties.
- Thermodynamics and Phase Stability: The model computes Gibbs free energies and phase diagrams, enabling efficient analysis of material stability under varying conditions.
- Mechanical Properties: MatterSim accurately predicts properties like bulk modulus, offering critical insights for engineering applications.
- Phonon Predictions: The model simulates lattice vibrations, which is critical for understanding thermal conductivity and dynamic stability.
- Molecular Dynamics: MatterSim is a reliable surrogate for first-principles methods, simulating materials under extreme temperatures and pressures.
MatterSim models also serve as a customization platform. Researchers can fine-tune the model using domain-specific data, reducing data requirements by up to 97%. For example, fine-tuning MatterSim models for water simulation at a higher theoretical level required only 3% of the data needed to train a similar model from scratch.
MatterSim models outperform universal force fields on datasets like MPF-TP, achieving superior accuracy in predicting materials’ energies, forces, and stresses. The model’s ability to simulate molecular dynamics across 118 diverse systems underscores its robustness and adaptability. For applications requiring high precision, MatterSimV1-5M delivers exceptional results. The model maintains over 90% success rates in simulations involving high temperatures and pressures, demonstrating robustness even in extreme conditions. The model’s pretraining on a vast dataset of 17 million structures ensures broad compositional and configurational coverage. This extensive training allows MatterSim to excel in tasks like materials discovery, where it identified thousands of stable structures not present in existing databases.
In conclusion, MatterSimV1-1M and MatterSimV1-5M combine the precision of first-principles methods with the efficiency of machine learning. These models enable researchers to simulate and predict material properties with unprecedented accuracy and speed. With applications ranging from material discovery to phase diagram construction, MatterSim models empower scientists to tackle complex materials design and engineering challenges. Researchers can access the models on GitHub, leveraging this cutting-edge tool to accelerate discoveries and what is possible in atomistic simulations.
Check out the GitHub Page and Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. If you like our work, you will love our newsletter.. Don’t Forget to join our 60k+ ML SubReddit.
The post Microsoft Released MatterSimV1-1M and MatterSimV1-5M on GitHub: A Leap in Deep Learning for Accurate, Scalable, and Versatile Atomistic Simulations Across Materials Science appeared first on MarkTechPost.