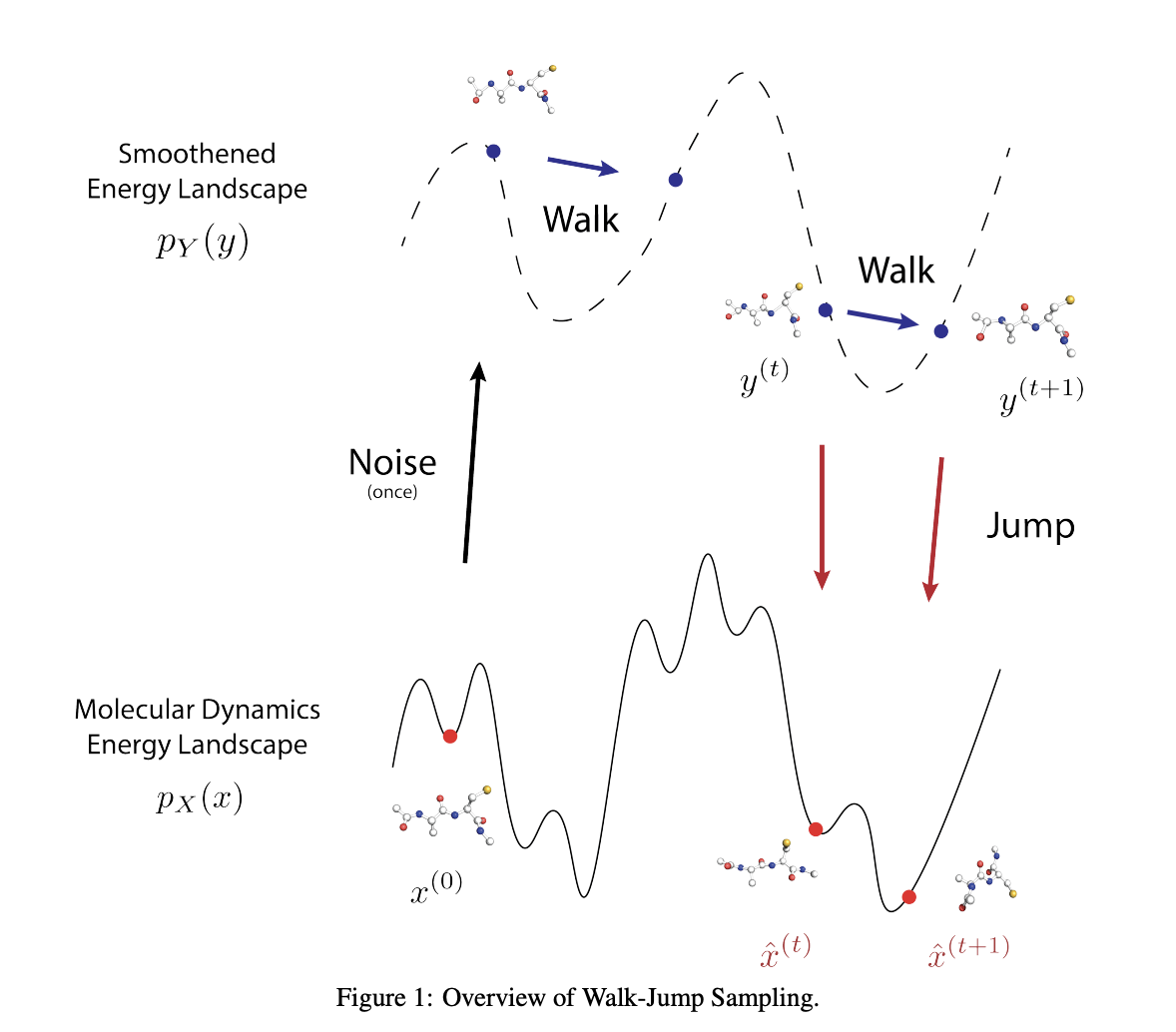
The dynamics of protein structures are crucial for understanding their functions and developing targeted drug treatments, particularly for cryptic binding sites. However, existing methods for generating conformational ensembles are plagued by inefficiencies or lack of generalizability to work beyond the systems they were trained on. Molecular dynamics (MD) simulations, the current standard for exploring protein movements, are computationally expensive and limited by short time-step requirements, making it difficult to capture the broader scope of protein conformational changes that occur over longer timescales.
Researchers from Prescient Design and Genentech have introduced JAMUN (walk-Jump Accelerated Molecular ensembles with Universal Noise), a novel machine-learning model designed to overcome these challenges by enabling efficient sampling of protein conformational ensembles. JAMUN extends Walk-Jump Sampling (WJS) to 3D point clouds, which represent protein atomic coordinates. By utilizing a SE(3)-equivariant denoising network, JAMUN can sample the Boltzmann distribution of arbitrary proteins at a speed significantly higher than traditional MD methods or current ML-based approaches. JAMUN also demonstrated a significant ability to transfer to new systems, meaning it can generate reliable conformational ensembles even for protein structures that were not part of its training dataset.
The proposed methodology is rooted in the concept of Walk-Jump Sampling, where noise is added to clean data, followed by training a neural network to denoise it, thereby allowing a smooth sampling process. JAMUN utilizes Langevin dynamics for the ‘walk’ phase, which is already a standard approach in Molecular dynamics MD simulations. The ‘jump’ step then projects back to the original data distribution, decoupling the process from starting over each time as is typically done with diffusion models. By decoupling the walk and jump steps, JAMUN smooths out the data distribution just enough to resolve sampling difficulties while retaining the physical priors inherent in MD data.
JAMUN was trained on a dataset of molecular dynamics simulations of two amino acid peptides and successfully generalized to unseen peptides. Results show that JAMUN can sample conformational ensembles of small peptides significantly faster than standard MD simulations. For instance, JAMUN generated conformational states of challenging capped peptides within an hour of computation, while traditional MD approaches required much longer to cover similar distributions. JAMUN was also compared against the Transferable Boltzmann Generators (TBG) model, showcasing a remarkable speedup and comparable accuracy, although it was limited to Boltzmann emulation rather than exact sampling.
JAMUN provides a powerful new approach to generating conformational ensembles of proteins, balancing efficiency with physical accuracy. Its ability to generate ensembles much faster than MD while maintaining reliable sampling makes it a promising tool for applications in protein structure prediction and drug discovery. Future work will focus on extending JAMUN to larger proteins and refining the denoising network for even faster sampling. By leveraging Walk-Jump Sampling, JAMUN offers a significant step towards a generalizable, transferable solution for protein conformational ensemble generation, crucial for both biological understanding and pharmaceutical innovation.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. If you like our work, you will love our newsletter.. Don’t Forget to join our 50k+ ML SubReddit.
[Upcoming Live Webinar- Oct 29, 2024] The Best Platform for Serving Fine-Tuned Models: Predibase Inference Engine (Promoted)
The post JAMUN: A Walk-Jump Sampling Model for Generating Ensembles of Molecular Conformations appeared first on MarkTechPost.